
Conocer la estructura electrónica molecular de las proteínas, así como su geometría espacial, es fundamental para la comprensión de los procesos químicos y biológicos de estos elementos esenciales para la vida. Un reciente trabajo, realizado por un grupo del departamento de Química Física Aplicada de la Universidad Autónoma de Madrid (UAM) (España), ha combinado cálculos teóricos y resultados experimentales que facilitan la determinación estructural de las proteínas.
Las funciones biológicas de una proteína están determinadas en gran parte por su estructura molecular, es decir, por la disposición espacial de sus átomos. Si bien en muchos casos las disposiciones geométricas de los átomos en las proteínas son estudiadas en estado sólido utilizando técnicas de rayos X, para condiciones cercanas a las fisiológicas resulta más apropiada la técnica de espectroscopía de resonancia magnética nuclear (RMN).
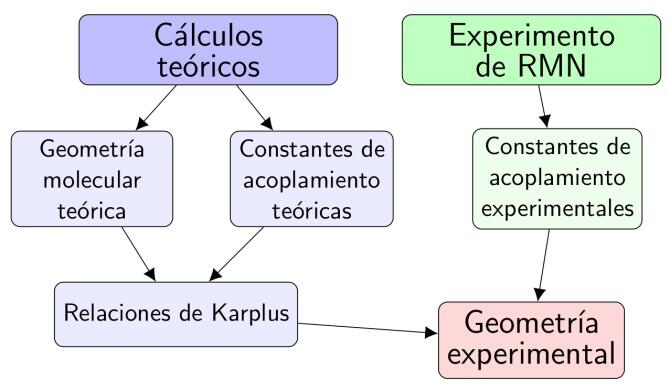
A partir de un espectro de RMN se obtienen parámetros importantes para la determinación estructural, como son las constantes de acoplamiento vecinales (3J). Estas constantes se deben a la interacción entre dos núcleos X···Y, que se comportan como si fueran dos diminutos imanes debido a sus espines.
Los valores de 3JXY reflejan la interacción entre los espines de núcleos vecinos, separados por tres enlaces (X-C-C-Y), interacción que se transmite a través de los electrones de los enlaces y que depende de la estructura molecular.
En un proceso inverso, la determinación de estos parámetros experimentales sirve para la determinación de la geometría. Sin embargo, para ello se necesita conocer las ecuaciones que relacionan los valores de 3JXY con la geometría de la molécula, a través de la dependencia con el ángulo diedro X-C-C-Y (ecuaciones de Karplus). Y es aquí donde los resultados teóricos son necesarios.
El nivel de los cálculos teóricos actuales permite la obtención de constantes de acoplamiento con exactitud comparable a las obtenidas directamente del experimento de RMN.
El trabajo, publicado en Journal of Chemical Theory and Computation, estudia la posibilidad de combinar métodos teóricos junto con resultados experimentales para determinar la disposición geométrica de los sustituyentes de las cadenas laterales de la proteína que dependen de la rotación alrededor de uno o varios enlaces simples. Con este propósito, los autores diseñaron un protocolo computacional a partir de modelos de dipéptidos.
Los cálculos teóricos rigurosos en este tipo de sistemas son muy costosos y por ello son necesarias aproximaciones que faciliten la obtención de resultados con la mayor exactitud posible y con un coste computacional asumible. Por este motivo, se han estudiado diferentes métodos o aproximaciones teóricas para encontrar los más apropiados para la determinación de las propiedades requeridas.
La utilización de métodos teóricos presenta algunas ventajas al ser un estudio multivariacional: por un parte, los valores experimentales de 3J dependen de numerosos factores, pudiéndose fijar algunas de las variables, obteniendo así la dependencia con las restantes; y por otra parte, los resultados obtenidos permiten hacer una estimación de la incertidumbre. (Fuente: UAM)








