
Investigadores del Consejo Superior de Investigaciones Científicas (CSIC) (España) que trabajan en el Instituto de Biomedicina de Valencia han llevado a cabo un estudio acerca de los determinantes genómicos de la especiación y propagación de la tuberculosis.
Los resultados del trabajo, en el que también ha participado el Instituto de Biología Integrativa de Sistemas (I2SysBio), centro mixto del CSIC y la Universitat de València, y la Fundación para el Fomento de la Investigación Sanitaria y Biomédica (FISABIO) de la Comunitat Valenciana, amplían los conocimientos sobre la evolución de las bacterias causantes de la tuberculosis en animales y humanos, y aparecen publicados en la revista Science Advances.
La tuberculosis es una enfermedad infecciosa causada por la bacteria Mycobacterium tuberculosis, que provoca una mortalidad devastadora en humanos y animales, y que también conlleva importantes pérdidas económicas. Conocer cómo se diferencian los distintos linajes bacterianos aumenta nuestra comprensión de los orígenes de la bacteria que causa la enfermedad y los mecanismos genéticos involucrados.
Los investigadores del CSIC en el Instituto de Biomedicina de Valencia Iñaki Comas y Álvaro Chiner Oms explican que “para conocer los eventos genómicos poblacionales que condujeron a la aparición del patógeno de la tuberculosis, hemos trabajado con el Complejo Mycobacterium tuberculosis o MTBC, que comprende un grupo de micobacterias conformado por Mycobacterium tuberculosis y Mycobacterium africanum, que afectan a los humanos, así como una serie de patógenos aislados de otras especies de mamíferos conocidos como Mycobacterium bovis, Mycobacterium pinnipedii, Mycobacterium orygis y Mycobacterium microti, entre otros”.
La creciente disponibilidad de datos genómicos poblacionales ha permitido una mejor comprensión de la diferenciación genotípica y ecológica entre bacterias estrechamente relacionadas. Esto ha permitido desarrollar modelos teóricos de cómo emergen las especies de bacterias y las regiones genómicas implicadas.
“Este estudio aplica por primera vez estos modelos a un patógeno que afecta a los humanos, para ello se han estudiado las bacterias más estrechamente relacionadas con MTBC, conocidas como Mycobacterium canettii o MCAN, una cepa aislada del Cuerno de África. Nuestro análisis confirma la hipótesis de que ambas compartieron un acervo genético común.
Además, hemos aprovechado la disponibilidad de secuencias genómicas de miles de cepas clínicas del complejo MTBC, así como de parientes cercanos como MCAN, para identificar nuevos determinantes genómicos en la aparición y posterior propagación del MTBC”, añade Iñaki Comas.
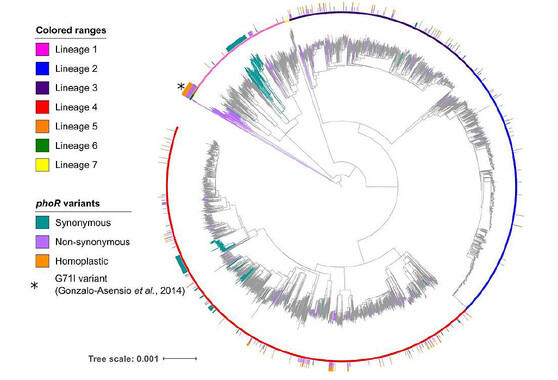
Los investigadores del Instituto de Biomedicina de Valencia han identificado el gen phoR como clave de un sistema involucrado en la virulencia, y que desempeñó un papel fundamental en la evolución del Complejo Mycobacterium tuberculosis.
“Trabajos anteriores habían mostrado que las mutaciones de phoR desempeñaron un papel central en la adaptación del patógeno a diferentes especies hospedadoras. Nosotros hemos demostrado la vinculación del gen phoR con la propagación temprana de la tuberculosis humana, así como en expansiones posteriores. Nuestro trabajo también demuestra que el estudio de la evolución de los patógenos ayuda a comprender los determinantes de su virulencia pasados y presentes”, concluye Comas.
En este trabajo también han participado investigadores del CIBER de Epidemiología y Salud Pública de Valencia, de la Universidad de Oslo, de la Universidad de Helsinki, del Swiss Tropical and Public Health Institute de Suiza, de la Universidad de Basel, del Microbiotica BioData Innovation Centre de Reino Unido, y del Francis Crick Institute de Reino Unido. (Fuente: CSIC)








